遺傳模式
- 一群體染色體隱性(autosomal recessive)遺傳疾病,共七種不同酵素/蛋白缺陷。
- 最常見類型為 CYP21A2 基因突變所造成的 21-hydroxylase deficiency(21-OHD),約占所有病例的 95%。
Steroidogenesis 機轉參考圖
腎上腺皮質以 cholesterol 為起點,分別合成 mineralocorticoid(aldosterone)、glucocorticoid(cortisol) 與 androgen 三大路徑。圖中以紅色 ✗ 標出 21-hydroxylase(P450c21) 缺損步驟,可見前驅物累積並轉往 androgen。
圖示重新繪製自 Brook's Clinical Pediatric Endocrinology, 7th edition, Chapter 9(簡化示意,未列出全部中間產物;Δ4 與 backdoor pathway 均可將累積之 17-OHP 導向 androgen)。
特徵與機轉
- 腎上腺類固醇生成途徑中的酵素缺陷,導致 cortisol 合成受損。
- cortisol 減少 → 下視丘–腦下垂體–腎上腺軸(HPA axis)的負回饋降低 → ACTH 分泌增加;ACTH 持續刺激腎上腺造成 adrenal hyperplasia,同時使上游 steroid precursors 累積。
以最常見的 21-hydroxylase(P450c21) 為例,它同時是 cortisol 與 aldosterone 合成的關鍵酵素,缺損時會在其上游造成 17-OHP/progesterone 累積並轉往 androgen。
目錄(共七種)
- Congenital lipoid adrenal hyperplasia (StAR)
- P450scc deficiency (CYP11A1)
- 21-hydroxylase deficiency (CYP21A2) ← 最常見
- 11β-hydroxylase deficiency (CYP11B1)
- 3β-HSD deficiency (HSD3B2)
- 17α-hydroxylase deficiency (CYP17A1)
- P450 oxidoreductase deficiency (POR)
| Condition | Gene | Protein | OMIM | Chromosome | Inheritance | Action | Associated features |
|---|---|---|---|---|---|---|---|
| Congenital lipoid adrenal hyperplasia | STAR | StAR | 201710 | 8p11.23 | AR | Steroidogenic enzyme | 46,XY DSD;impaired gonadal steroidogenesis |
| P450 side-chain cleavage insufficiency | CYP11A1 | P450scc | 613743 | 15q24.1 | AR | Steroidogenic enzyme | 46,XY DSD;impaired gonadal steroidogenesis |
| 3β-Hydroxysteroid dehydrogenase type II insufficiency | HSD3B2 | 3β-HSD2 | 201810 | 1p12 | AR | Steroidogenic enzyme | 46,XY DSD;impaired gonadal steroidogenesis;46,XX clitoromegaly |
| 17α-Hydroxylase / 17,20-lyase insufficiency | CYP17A1 | P450c17 | 202110 | 10q24.32 | AR | Steroidogenic enzyme | 46,XY DSD;impaired gonadal steroidogenesis;hypertension |
| P450 oxidoreductase | POR | P450 oxidoreductase | 201750 | 7q11.23 | AR | Steroidogenic enzyme | Antley–Bixler syndrome(craniosynostosis, skeletal features, choanal atresia);atypical genitalia(46,XY 與 46,XX);impaired gonadal steroidogenesis at puberty |
| 21-Hydroxylase deficiency | CYP21A2 | P450c21 | 201910 | 6p21.33 | AR | Steroidogenic enzyme | 46,XX DSD;virilization, early puberty |
| 11β-Hydroxylase deficiency | CYP11B1 | P450c11 | 202010 | 8q24.3 | AR | Steroidogenic enzyme | 46,XX DSD;virilization, early puberty, hypertension |
表格重新繪製自 Brook's Clinical Pediatric Endocrinology, 7th edition, Chapter 9。
21-hydroxylase deficiency(個論)
提醒:以下個論以最常見的 21-OHD 為主,其他六種 CAH 的臨床表現不完全相同(詳見上方比較表與併發症整理)。
① 失鹽型(Salt-wasting CAH)
- 約占經典型的 75%;21-hydroxylase 活性嚴重降低或完全缺乏 → cortisol ↓、aldosterone ↓、androgens ↑↑。
- 新生兒可能發生 salt-wasting crisis:低血鈉、高血鉀、代謝性酸中毒、低血容量、低血壓。
- 電解質異常多於出生後 第 1–2 週出現,第 7–14 天風險最高;若未治療,約 75% 於出生後 2–3 週內發生危及生命的鹽流失危象。
- 46,XX 新生兒常出現不同程度的外生殖器男性化(virilized external genitalia)。
② 單純雄性化型(Simple Virilizing CAH)
- 約保留 1–10% 的 21-hydroxylase 活性;aldosterone 大致足夠,通常無明顯鹽流失;cortisol 不足、androgen 明顯增加。
- 外生殖器男性化(virilization);若未即時治療 → 出生後生長加速、性早熟、骨齡提前(premature epiphyseal closure)→ 最終成人身高矮小。
- 重大生理壓力(發燒感染、trauma、手術)下,仍可能出現輕度或暫時性鹽分流失。
③ 非典型(Nonclassic CAH)
- 約保留 20–80% 的 21-hydroxylase 活性;多於兒童期晚期、青春期或成年期才出現症狀。
- 主要表現為 androgen excess:precocious pubarche、hirsutism、oligomenorrhea、acne、女性 subfertility。
| Mutation | Enzyme activity(% of wild type) | Most common associated phenotype |
|---|---|---|
| Gene deletion | 0 | Classic |
| Large gene conversion | 0 | Classic |
| p.Pro31Leu | 30–60 | Non-classic |
| c.293-13A>G | 1 | Classic |
| c.293-13C>G | 1 | Classic |
| p.Gly111ValfsTer21 | 0 | Classic |
| p.Ile173Asn | 3–10 | Classic / simple virilizing |
| p.Ile237Asn / Val238Glu / Met240Lys | 0 | Classic |
| p.Val282Leu | 20 | Non-classic |
| p.Leu308PhefsTer6 | 0 | Classic |
| p.Gln319Ter | 0 | Classic |
| p.Arg340His | 20–50 | Non-classic |
| p.Arg357Trp | 2 | Classic / simple virilizing |
| p.Pro454Ser | 20–50 | Non-classic |
Table 9.6, Brook's Clinical Pediatric Endocrinology, 7th edition, Chapter 9。
NB:codon 編號可能與早期文獻不同(例:p.Ile173Asn 舊稱 p.Ile172Asn;p.Val282Leu 舊稱 p.Val281Leu)。
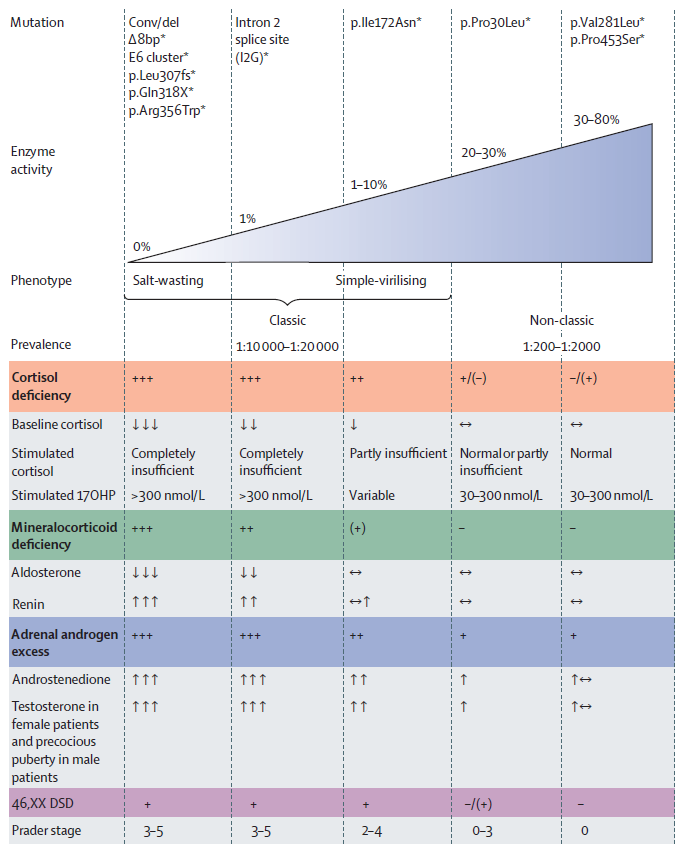
來源:Auer MK, et al. Congenital adrenal hyperplasia. Lancet. 2023 Jan 21;401(10372):227–244. doi:10.1016/S0140-6736(22)01330-7
台大醫院(NTUH)先天性腎上腺增生症之臨床經驗:J Formos Med Assoc. 2018 Feb;117(2):126–131. doi:10.1016/j.jfma.2017.03.008
(可再補充本院世代之重點數據,例如診斷年齡、篩檢後成效、基因型分布等。)
不負責任併發症整理
教學速記用;下方先列六個常考問題,最後附一張整合對照矩陣方便一眼掃過。
| 酵素缺陷 | 17-OHP 篩得出 | 46,XX DSD | 46,XY DSD | 失鹽(↓Na ↑K) | 高血壓(↑Na ↓K) |
|---|---|---|---|---|---|
| StAR | — | — | ✓ | ✓ | — |
| P450scc(CYP11A1) | — | — | ✓ | ✓ | — |
| 3β-HSD(HSD3B2) | ✓ | ✓ | ✓ | ✓ | — |
| 17α(CYP17A1) | — | — | ✓ | — | ✓ |
| POR | ✓ | ✓ | ✓ | ✓ | — |
| 21-OHD(CYP21A2) | ✓ | ✓ | — | ✓ | — |
| 11β(CYP11B1) | ✓ | ✓ | — | — | ✓ |
備註:POR 之外生殖器異常於 46,XX 與 46,XY 皆可能發生;3β-HSD 亦同時影響兩者,故此二者在「46,XX/46,XY DSD」欄皆標記為 ✓。
新生兒篩檢(NB screening)
- 篩檢指標:17-羥基黃體酮(17-hydroxyprogesterone, 17-OHP)。
- 台灣新生兒篩檢 17-OHP cut-off 參考值:GA < 37 週:7.2 ng/mL;GA ≥ 37 週:5.8 ng/mL。
- 可被篩檢發現的 CAH 類型:21-hydroxylase deficiency、11β-hydroxylase deficiency、P450 oxidoreductase deficiency(PORD)、3β-HSD type 2 deficiency。
單位換算:17-OHP 分子量 330.4611 g/mol;臨床常用換算 血清(nmol/L)÷ 3 ≈ 新生兒篩檢血片(ng/mL)。
在地資料提醒:上列台灣篩檢 cut-off 依國家篩檢計畫/各篩檢中心而定,實際採用值請以最新公告為準。
理學檢查的重點
- Hyperpigmentation(POMC 在製造 ACTH 的過程中會同時產生 β-MSH,因而造成色素沉積)。
- Over-virilization(46,XX DSD):以 Prader staging 分級(見下)。
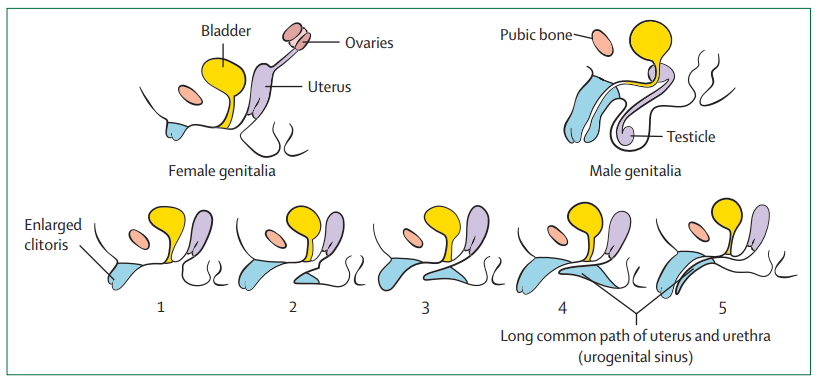
來源:Auer MK, et al. Lancet. 2023 Jan 21;401(10372):227–244. doi:10.1016/S0140-6736(22)01330-7
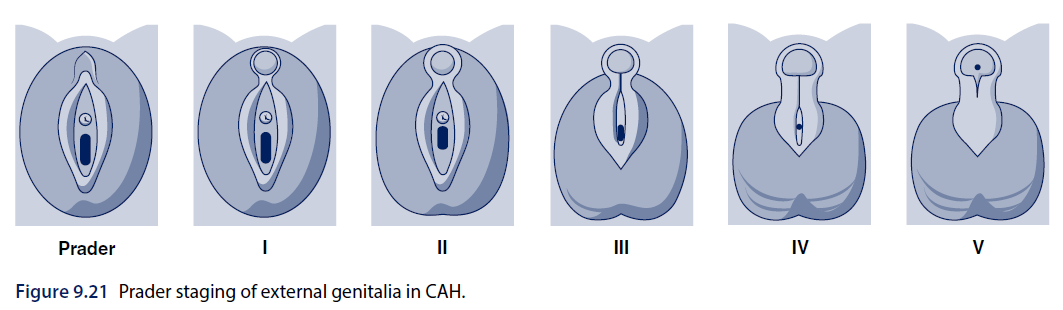
來源:Brook's Clinical Pediatric Endocrinology, 7th edition, Chapter 9(Figure 9.21)。
| Prader stage | Features |
|---|---|
| 1 | Mild clitoromegaly |
| 2 | Clitoromegaly, posterior labial fusion |
| 3 | Greater clitoromegaly, complete labial fusion, some 'scrotalization' of the labia, single perineal opening |
| 4 | Increased phallic size, complete labial fusion, 'scrotalization' of the labia, urethra-like opening at base or lower part of the phallus |
| 5 | Penis-like phallus, complete labial fusion, scrotal-like appearance of the labia, urethral meatus at the tip of the phallus |
表格重新繪製自 Brook's Clinical Pediatric Endocrinology, 7th edition, Chapter 9。
ACTH stimulation test(ACTH 刺激試驗)
- 目標:確認診斷及評估腎上腺類固醇生成能力。
- 標準方法:Synacthen®(合成 ACTH)0.25 mg IV,於給藥前及給藥後測量 17-OHP、cortisol 等激素濃度。
| 時間 | 紫頭管 2cc |
紅頭黃頂管 ×2 0分:10 或 8cc(5+5 / 4+4)、60分:5cc |
綠頭黑管 ×2(置冰) 5cc(2.5+2.5) |
1cc | 0.5cc | 小紫頭管 0.5cc |
小紫頭管 | 紫頭管 2cc(2.5+2.5) |
血量 | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ACTH | Cortisol | 17-OHP | Andro- stenedione |
Testos. | DHEAS | PRA | Aldo- sterone |
e- | gas | 血糖 | CBC | DNA | ||
| 0′ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | 17cc |
| 60′ | — | ✓ | ✓ | ✓ | ✓ | ✓ | — | — | — | — | — | — | — | 11cc |
採檢提醒:本表依原始採檢單重製(採檢管以管蓋顏色分組);實際採檢管別、血量與各時間點勾選請以貴單位最新流程為準。
| Classic 21-OHD | Non-classic 21-OHD | Unaffected | |
|---|---|---|---|
| Basal 17-OHP | >300 nmol/L (>10,000 ng/dL) | 6–300 nmol/L (200–10,000 ng/dL) | <6 nmol/L (<200 ng/dL) |
| Stimulated 17-OHP | >300 nmol/L (>10,000 ng/dL) | 31–300 nmol/L (1,000–10,000 ng/dL) | <30 nmol/L (<1,000 ng/dL) |
ACTH 刺激後 17-OHP > 30 nmol/L(1,000 ng/dL)支持 21-OHD 診斷;classic 通常 > 300 nmol/L(10,000 ng/dL),non-classic 介於 30–300 nmol/L。Auer MK, et al. Lancet. 2023;401:227–244. doi:10.1016/S0140-6736(22)01330-7
基因檢測(以 21-hydroxylase 為例)
- CYP21A2 基因:位於 chromosome 6,編碼 21-hydroxylase;大多數患者為 compound heterozygotes(攜帶兩個不同致病變異)。
- CYP21P(CYP21A1P)偽基因:與 CYP21A2 序列相似度高達 98%,為致病變異的重要來源。
- 常見致病機轉:Gene conversion(75%)、Gene deletion due to meiosis(20%)、Other mutations(5%)。
CAH-X 症候群(CAH-X Syndrome)
- CYP21A2 與 TNXB 基因部分重疊;當大片段缺失同時影響兩者 → 形成 contiguous gene syndrome,即 CAH-X syndrome(CAH + Ehlers-Danlos syndrome)。
- 臨床特徵:joint hypermobility、arthralgia、hernia、結締組織異常、structural heart anomalies。
- 盛行率:典型 CAH 約 15%;若帶有 30 kb deletion 可高達 30–60%。
- 建議:對帶有大型 CYP21A2 缺失者,考慮進一步評估 CAH-X syndrome 的可能。
傳統治療
典型 21-OHD 治療建立在兩大支柱:① glucocorticoid + mineralocorticoid 替代,以及 ② androgen 控制(以 cortisol 重建對 HPA 軸的負回饋,抑制過量 androgen)。
① Glucocorticoid 替代
- 兒童/青少年(至最終身高前)首選 hydrocortisone(對生長影響最小);每日約 8–15 mg/m²,一天分 3–4 次。青少年可到 10–17 mg/m²,但應避免 > 17 mg/m² 以免影響生長。
- 常需超生理劑量才能壓制 androgen,故 CAH 用量常高於一般腎上腺功能不全者,須留意過度暴露之後遺症。
- 幼兒可用 taste-masked HC 顆粒(Alkindi,台灣尚無)提供低劑量給藥。
- 成人達最終身高後,可考慮長效 GC(prednisolone/dexamethasone)改善順從性;但心血管代謝與骨質副作用較高,兒童不建議使用長效 GC。
② Mineralocorticoid 替代
- Fludrocortisone:每日約 0.05–0.2 mg(標準 0.1 mg);salt-wasting 型須終身補充。
- 嬰兒前 6 個月常需較高劑量(生理性 mineralocorticoid 阻抗);合併 NaCl 補充 1–2 g(17–34 mEq) 於出生第一年,對電解質與生長皆重要。
- 使用合成 GC(mineralocorticoid 活性較低)者,fludrocortisone 劑量可能需提高。
| 藥物 | 兒童每日劑量 | 頻次 | 備註 |
|---|---|---|---|
| Hydrocortisone (cortisone acetate 為替代) | 嬰兒 8–12 mg/m²;兒童 10–15 mg/m²;青少年 10–17 mg/m² | 一天 3–4 次 | 生理性、短半衰期;最終身高與代謝/骨質長期結果最佳 |
| Fludrocortisone | 0.01–0.1 mg | 一天 1–2 次 | salt-wasting 型終身;嬰兒前 6 個月常需較高劑量 |
| Sodium chloride(NaCl) | 1–2 g(17–34 mEq) | 依餵食分次 | 出生第一年;夏季或大量流汗時額外補充 |
劑量整理自 Auer MK, et al. Congenital adrenal hyperplasia. Lancet. 2023 Jan 21;401(10372):227–244(Table 1). doi:10.1016/S0140-6736(22)01330-7
急性腎上腺危象與 sick day rules
什麼是腎上腺危象(adrenal crisis)?
- 臨床特徵:電解質失衡、對 catecholamine 無反應、因體液流失造成 hypovolemic shock,部分伴隨 hypoglycemia。
- 最常見誘因為 腸胃炎(gastroenteritis):口服 GC 吸收下降、同時壓力下需求上升。
- 流行病學:典型 CAH 兒童於出生前 4 年,約 1/5(約 20%)曾發生腎上腺危象;即使新生兒篩檢普及,仍屬潛在致命狀況。
Sick day rules(壓力劑量/dose escalation)
- 遇發燒感染、外傷、手術等生理壓力時,須提高 hydrocortisone 劑量(一般為平日的 2–3 倍)。
- 無法口服/嘔吐/嚴重疾病時:改以肌肉或靜脈注射 hydrocortisone(成人可 off-label 皮下注射),並補充葡萄糖與電解質。
- 病人與照顧者/伴侶都應:能辨識早期徵兆、熟悉劑量調整,並隨身攜帶緊急卡與注射型 hydrocortisone。
非典型 CAH 也要注意
- 非典型(non-classic)患者於嚴重疾病或大手術時,若 ACTH 刺激後 cortisol 反應不足(< 14–18 μg/dL,即 < 400–500 nmol/L),仍建議給予 stress dosing。
Auer MK, et al. Congenital adrenal hyperplasia. Lancet. 2023;401:227–244(Adrenal crisis). doi:10.1016/S0140-6736(22)01330-7
懷疑 crisis 的緊急情況:立刻通知 fellow,並留以下檢驗
- 1️⃣ Hyponatremia survey(spot urine Na、urine Osmo、serum Na、serum Osmo)。若診斷不確定,留到 urine 後再開始補 Na;若情況緊急,請立刻導尿!
- 2️⃣ ACTH、Cortisol
給藥
- 1️⃣ 先 bolus Solu-Cortef IV push:
• 25 mg:newborn 或 BSA < 0.25 m²
• 50 mg:preschool age 或 BSA 0.25–0.5 m²
• 100 mg:school age 或 BSA > 0.5 m² - 2️⃣ cIF(continuous infusion) Then Solu-Cortef 100 mg/m²/day continuous infusion
最好有獨立一條 line for Solu-Cortef infusion:將一天總量泡在 240 mL N/S、run 10 mL/hr,24 小時內連續滴注;若為 newborn 可將一天總量泡在 48 mL N/S、run 2 mL/hr。
CRH receptor antagonist
Crinecerfont 美國 FDA 核可
- 商品名 Crenessity;2024/12 美國 FDA 核可,用於 classic CAH(21-OHD)之 ≥ 4 歲兒童與成人,與 glucocorticoid 併用控制 androgen。
- 機轉:CRF1(CRH type 1)receptor antagonist,口服每日兩次;拮抗腦下垂體 CRF1 → 降低 ACTH → 減少 adrenal androgen,進而有機會下修 GC 劑量。
在地提醒:台灣 TFDA 藥證與健保給付狀態請自行確認,本工具不臆測。
| 研究 | 對象/設計 | 主要結果(vs 安慰劑) |
|---|---|---|
| CAHtalyst Adult | 182 位成人(≥ 18 歲) 24 週雙盲 RCT | 第 24 週 GC 劑量變化:crinecerfont −27.3% vs 安慰劑 −10.3%(維持 androstenedione 控制;LSMD 約 −17%,P < 0.0001)。 62.7% vs 17.5% 達成「生理性 GC 劑量且維持 androstenedione 控制」。 |
| CAHtalyst Pediatric | 103 位兒童(4–17 歲,2:1) 28 週雙盲 RCT | 達成「GC 劑量↓ 及/或 androstenedione↓」至少一項門檻:90% vs 21%。 |
臨床意義:可在維持 androgen 控制的同時下修 GC 劑量,減少長期超生理劑量 GC 的心血管代謝與骨質負擔。安全性:常見不良反應為 fatigue、headache,未見藥物相關 adrenal crisis。
兩年延伸數據(AACE 2026):成人平均 GC 由約 32 mg/天降至約 19.4 mg/天,約 70% 維持於生理範圍。
Auchus RJ, et al. Phase 3 Trial of Crinecerfont in Adult CAH. N Engl J Med. 2024;391(6):504–514. doi:10.1056/NEJMoa2404656
Sarafoglou K, et al. Phase 3 Trial of Crinecerfont in Pediatric CAH. N Engl J Med. 2024;391(6):493–503. doi:10.1056/NEJMoa2404655
緩釋型 hydrocortisone
Efmody 歐盟核可 英國核可
- 研發名 Chronocort;歐盟(2021/05)與英國 MHRA 核可用於治療 21-OHD 之 ≥ 12 歲青少年與成人。
- 採 delayed + sustained release 設計,2/3 劑量睡前給以模擬夜間 cortisol 上升與晨間高峰,改善荷爾蒙控制、可望減量。
- 2026 年起歐盟適應症已擴及 adrenal insufficiency。
在地提醒:美國、台灣之藥證狀態請自行確認。
其他發展中(pipeline)
- 其他非 GC 途徑:anti-ACTH monoclonal antibody、MC2R(ACTH receptor)antagonist(如 atumelnant,研究中)、antiandrogen、aromatase inhibitor、abiraterone acetate 等,皆以限制 adrenal androgen 過量為目標。
- Gene therapy 與 stem-cell therapy 則以修復缺損的類固醇生成為方向。
新興治療整理自 Auer MK, et al. Lancet. 2023;401:227–244(Figure 4)doi:10.1016/S0140-6736(22)01330-7;藥證狀態參 FDA(Crenessity, 2024/12)與 EMA/MHRA(Efmody)核可公告。
CAH 的主要爭議集中在女性男性化外生殖器的處置:包括產前預防男性化與出生後手術矯正,兩者皆具高度爭議。
生殖器手術(Genital surgery)
- 針對 46,XX 男性化外生殖器的早期手術矯正特別具爭議。基於部分不理想的手術結果,intersex 病友倡議團體主張:手術應在個案能自主同意(informed consent)時才進行。
- 多份國際人權文件(如 2013 UN 特別報告、2017 歐洲理事會 Resolution 2191)主張:非攸關健康之手術應延後至個案能自主決定,並提供充分心理支持。
- 長期追蹤顯示,即使使用現代手術技術,部分患者在性功能與陰蒂敏感度結果仍不理想;有 meta-analysis 報告陰蒂敏感度下降、陰道狹窄與性交不適。保留敏感度的新式術式與腹腔鏡已被報導,但需更長時間評估。
- 一項小型研究顯示:Prader 3–4 女童經 glucocorticoid 治療後,陰蒂長度可縮短至出生時的一半以下,提示延後手術對部分家庭可能可接受,但仍強調嚴格控制與青春期心理支持。
產前 dexamethasone(Prenatal treatment)
- 自 1984 年起使用,於孕早期給予 dexamethasone(可通過胎盤),以預防女胎外生殖器男性化。
- 倫理困境:須在能基因型判定前(< 7 週)就開始;即使 5–6 週可先排除男胎,所有女胎仍會被治療至確診。未受影響的女胎會被不必要地暴露至第一孕期末。
- 安全性未明:對胎兒腦部結構、神經認知與代謝之影響研究樣本多偏小、結論不一;母體副作用(水腫、體重增加、striae、睡眠與情緒變化)常見但多可逆。
- 目前建議:僅在完整揭露風險、經 IRB 核准下使用,並納入長期追蹤;國際間使用差異大。
Auer MK, et al. Congenital adrenal hyperplasia. Lancet. 2023;401:227–244(Controversial therapies). doi:10.1016/S0140-6736(22)01330-7
症狀與理學檢查
- 月經是否規則
- 多毛症(hirsutism)
- 色素沉著(hyperpigmentation)
- 類庫欣症表現(cushingoid features)
兒童與青少年應定期追蹤
- 生長曲線(growth chart)
- 骨齡(bone age)骨齡延遲(bone age delay)不一定代表需減少藥物劑量,應結合臨床狀況及實驗室數據綜合判讀。
- Tanner stage
其他監測項目
- 血壓(blood pressure)
- 身體質量指數(BMI)
- 骨密度檢查(DXA)
實驗室監測(Laboratory Monitoring)
| Marker | 反映/意義 | 目標與注意事項 |
|---|---|---|
| 17-OHP | 短期控制情形 | 受 ACTH diurnal variation 影響,應晨間抽血;女性最佳為濾泡期(黃體期自然升高)。目標維持於正常上限 ULN ~ < 36 nmol/L;完全正常值可能代表過度治療 |
| Androstenedione | 較長期的 androgen 控制狀態 | 維持於正常範圍內 |
| Testosterone (女性患者) | androgen 控制 | 可測 total testosterone 或計算 free testosterone |
| Progesterone | androgen/懷孕控制 | 尤其適用有懷孕計畫之女性;目標 follicular phase < 0.6 ng/mL(< 2 nmol/L) |
| Gonadotropins (LH/FSH) | 男性 androgen 控制 | 男性患者 LH、FSH 不應受到抑制;若被抑制可能表示 androgen 過量 |
| DHEAS | — | 使用 glucocorticoid 後通常下降,不適合作為疾病控制監測指標 |
| Electrolytes | 失鹽狀態 | salt-wasting CAH 患者監測 serum sodium、potassium |
| Plasma Renin | mineralocorticoid 補充是否足夠 | 通常以維持在正常值上限(upper normal range)為治療目標 |
監測原則整理自 Auer MK, et al. Lancet. 2023;401:227–244(Table 2). doi:10.1016/S0140-6736(22)01330-7
相關腫瘤(控制不佳者)
- Adrenal myelolipoma
- Adrenal adenoma
- 睪丸腎上腺殘餘組織腫瘤(Testicular Adrenal Rest Tumor, TART)多為雙側、良性;與 androgen 控制不佳及 ACTH 上升相關。可能導致睪丸功能受損、不孕症;以超音波監測,若偵測到可提供精子冷凍保存。
長期預後:生育(Fertility)
女性
- steroid 荷爾蒙失衡可致 30–60% 未使用避孕藥女性出現月經異常;升高的 17-OHP/progesterone 具 progestogen 效應,干擾子宮內膜與受孕。
- 但在適當 GC 替代下,多數仍可懷孕、fecundity 不受明顯影響;備孕時常需提高 GC(bedtime prednisolone 效果常優於 hydrocortisone),把 progesterone 維持 < 2 nmol/L。
男性與 TART
- 男性有 hypogonadism/infertility 風險;成因包括 androgen 過量抑制 HPG 軸,及 睪丸腎上腺殘餘組織(TART)。
- TART 多為雙側、良性;平均見於約 37% 典型 CAH 男性,隨年齡與基因型嚴重度增加,常與 androgen 控制不佳/ACTH 上升相關。
- 加強 GC 抑制可能使 TART 縮小、改善精蟲生成;手術無法改善性腺功能,僅在腫瘤造成不適或疼痛時考慮;偵測到 TART 可提供精子冷凍保存。
心血管代謝與骨質健康
- 心血管代謝:心血管疾病罹病率增加;常見血壓上升、動脈硬化、胰島素阻抗;肥胖率可達 40%;不少患者睡眠期收縮壓下降不足(已知心血管風險因子)。
- 骨質:lumbar spine 與 femoral neck 的 Z score 較低;合成 GC 對骨質的負面影響大於 hydrocortisone;男女骨折風險皆增加。
- 長期超生理劑量 GC、肥胖、胰島素阻抗與 androgen 過量交互作用,共同推高血壓與心血管風險——這也是新藥(Crinecerfont)減量的重要動機。
Transition of care(照護轉銜)
- 由兒科轉銜至成人專科,是持續規則追蹤、減少長期後遺症的基礎。
- 青少年應逐步熟悉自身疾病、治療策略與腎上腺危象處理,並持續獲得心理支持;生育與性健康議題應納入衛教。
- 一般認為 16–20 歲 為適當的轉銜時機。
Auer MK, et al. Congenital adrenal hyperplasia. Lancet. 2023;401:227–244(Long-term outcomes;Transition of care). doi:10.1016/S0140-6736(22)01330-7
提醒:17-OHP 為良好診斷指標,但非理想的治療達標指標——把 17-OHP 壓到完全正常常代表 GC 過度治療。臨床應綜合生長速度、骨齡、androstenedione 等一併判讀。
QCAH 會遺傳嗎?下一胎的風險有多少?
Q為什麼藥要一天吃好幾次?生病時還要加量?
Q女寶寶外觀比較男性化,之後會改善嗎?一定要開刀嗎?
Q長期吃類固醇會不會有副作用?
Q平常要準備什麼以因應緊急狀況?
以上為一般性衛教,個別狀況請與您的主治醫師討論。